Content from Command line tools for quality control
Last updated on 2024-09-08 | Edit this page
Overview
Questions
- How to perform quality control of NGS raw data?
Objectives
- Assess short reads FASTQ quality using
FASTQE - Compare reads before and after quality filtering with
fastp
Introduction
During sequencing, the nucleotide bases in a DNA or RNA sample (library) are determined by the sequencer. For each fragment in the library, a sequence is generated, also called a read, which is simply a succession of nucleotides.
Modern sequencing technologies can generate a massive number of sequence reads in a single experiment. However, no sequencing technology is perfect, and each instrument will generate different types and amount of errors, such as incorrect nucleotides being called. These wrongly called bases are due to the technical limitations of each sequencing platform.
Therefore, it is necessary to understand, identify and exclude error-types that may impact the interpretation of downstream analysis. Sequence quality control is therefore an essential first step in your analysis. Catching errors early saves time later on.
To take a look at sequence quality along all sequences, we can use FASTQE. It is an open-source tool that provides a simple and fun way to quality control raw sequence data and print them as emoji. You can use it to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis.
We will also use the tool fastp which can be used to
filter sequence data to remove low quality reads.
OUTPUT
fastqe 1.0Challenge 2: Can you do the same for
fastp? Hint: try -v as well as
--version
Callout
When using the command line code, lines that start with the # sign are typically text descriptors or instructions, not lines of code.
The # character is used to indicate comments. A line starting with # line doesn’t do anything, in this case it’s just telling you what the next line of code does which is navigate to your computer’s desktop - We will stick with this convention throughout the activity - Comments can occur anywhere in a line, anything after the # will be ignored
OUTPUT
$ fastp --version
fastp 0.20.1You should get the same result from using -v and
--version which are known as short and long options
respectivlely. Long options will typically (but not always) have a
corresonidng short option.
Command line structure
We have mentioned commands and options so far. The syntax is typically as follows:

OUTPUT
dev/ home/ proc/ shared/ tmp/This is the filesystem of our browser-based portal.
Case Study
Here’s brief background on the in class metagenomics project that we’re collecting sequencing data for. Garter snakes excrete sexually dimorphic pheromones to attract a mate. The hypothesis of our experiment is that male and female garter snakes host unique microbial communities in their mouths, cloacae and musk glands that contribute to sexually dimorphic bioengineering of these pheromone molecules. Figure 1 provides an overview of our 16S metagenomics analysis pipeline. For this lesson though, all you need are the FASTQ files.
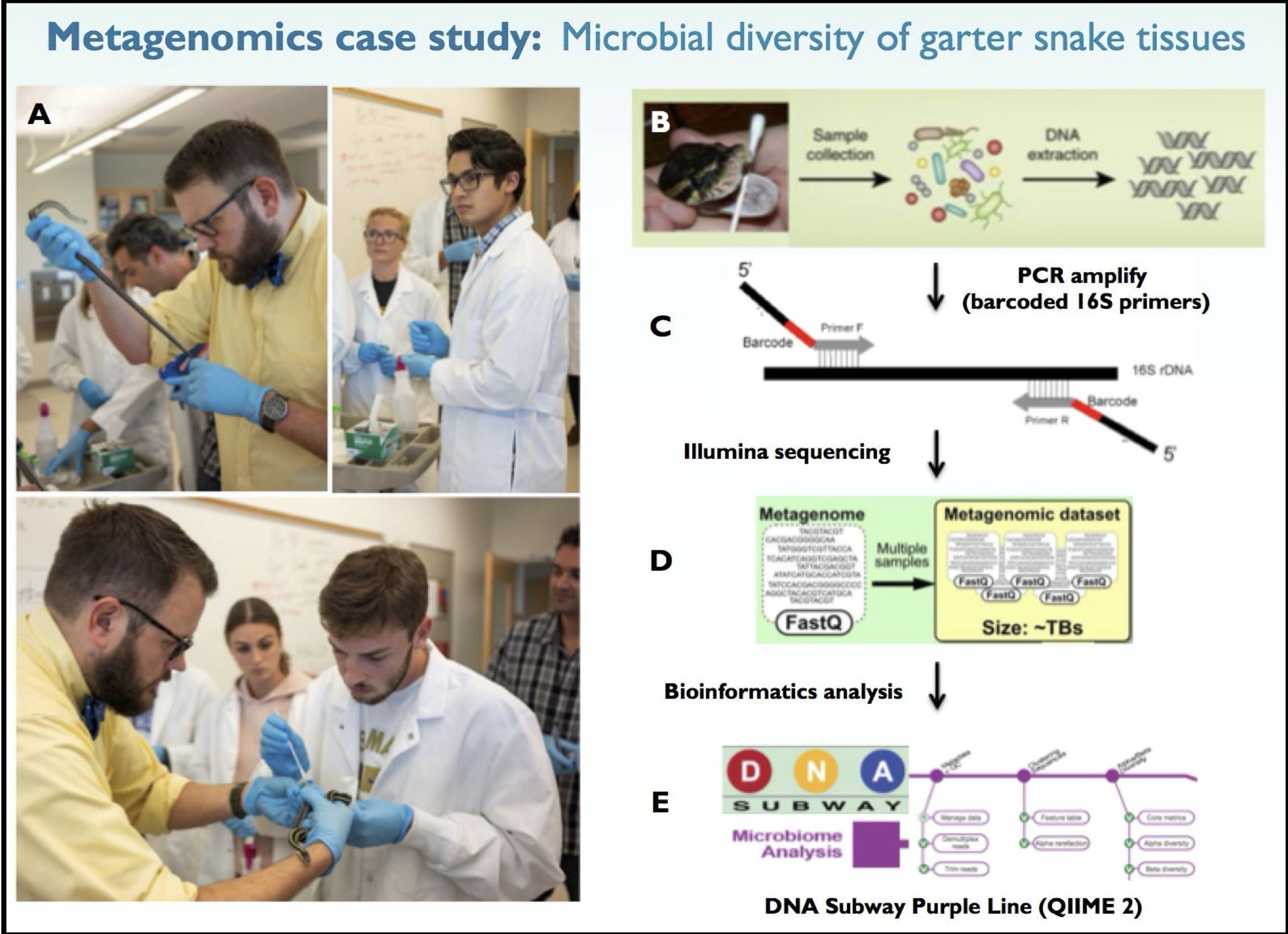
The data for this experiment is located in the
/shared/fastqe/ folder.
Challenge 4: What are the files available from this experiment?
Hint: Use the ls command we have already seen
Key Points
- Options can be long and short
- Structure of a shell command
- We have confirmed
fastpandfastqeare installed - The case study and location of the data
Content from The FASTQ format and FASTQE
Last updated on 2024-09-08 | Edit this page
Introduction
The first step of any Next Generation Sequencing (NGS) analysis pipeline is checking the quality of the raw sequencing reads in each FASTQ formatted file. If the sequence quality is poor, then your resulting downstream analysis will be inaccurate and misleading. FastQC is a popular software used to provide an overview of basic quality metrics for NGS data. In this lesson, you will use an even more universal form of communication to analyze FASTQ files, THE EMOJI.
The tool we will use for this visualisation is
fastqe
If you have fastqe installed correctly, you should get
usage information:
OUTPUT
$ fastqe --help
Read one or more FASTQ files, compute quality stats for each file, print as emoji... for some reason.😄
🚨 Rust and WebAssembly beta: only command line options with a ✅ are functional
rustc 1.75.0-beta.7 emcc 3.1.50
Usage: fastqe [OPTIONS] [FASTQ_FILE]...
Arguments:
[FASTQ_FILE]... Input FASTQ files
Options:
--noheader Hide the header before sample output ❌
--output <OUTPUT_FILE> Write output to OUTPUT_FILE instead of stdout
--long <READ_BUFFER> Buffer memory for long reads up to READ_BUFFER bp long ❌ [default: 500]
--log <LOG_FILE> Record program progress in LOG_FILE ✅
-h, --help Print help
-V, --version Print version
Emoji options:
--bin Use binned scores (🚫 💀 💩 🚨 😄 😆 😎 😍 ) ❌
--custom <CUSTOM_DICT> Use a mapping of custom emoji to quality in CUSTOM_DICT (🐍🌴) ❌
--noemoji Use mapping without emoji (▁▂▃▄▅▆▇█) ❌
Statistics options:
--minlen <N> Minimum length sequence to include in stats ✅ [default: 0]
--scale Show relevant scale in output ❌
--nomean Hide mean quality per position ❌
--min Show minimum quality per position ❌
--max Show maximum quality per position ❌
HTML report options:
--html Output all data as html ❌
--window <W> Window length to summarise reads in HTML report ❌ [default: 1]
--html_escape ❌ Escape html within output, e.g. for Galaxy parsing
For more information, vist https://fastqe.comFASTQE has a lot of options! Note that this version does not implement all of them, however we will be using it in the default mode.
The FASTQ format
Although it looks complicated (and maybe it is), the FASTQ format is easy to understand with a little decoding.
Each read, representing a fragment of the library, is encoded by 4 lines:
- Always begins with @ followed by the information about the read
- The actual nucleic sequence
- Always begins with a + and contains sometimes the same info in line 1
- Has a string of characters which represent the quality scores associated with each base of the nucleic sequence; must have the same number of characters as line 2
So for example, the first sequence in our file is:
@M00970:337:000000000-BR5KF:1:1102:17745:1557 1:N:0:CGCAGAAC+ACAGAGTT
GTGCCAGCCGCCGCGGTAGTCCGACGTGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACCGAAGAACATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAAAAAACAAAAAAAAAAAAAGAAGCAAATGACGATTCAAGAAAGAAAAAAACACAGAATACTAACAATAAGTCATAAACATCATCAACATAAAAAAGGAAATACACTTACAACACATATCAATATCTAAAATAAATGATCAGCACACAACATGACGATTACCACACATGTGTACTACAAGTCAACTA
+
GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGFGGGGGGAFFGGFGGGGGGGGFGGGGGGGGGGGGGGFGGG+38+35*311*6,,31=******441+++0+0++0+*1*2++2++0*+*2*02*/***1*+++0+0++38++00++++++++++0+0+2++*+*+*+*+*****+0**+0**+***+)*.***1**//*)***)/)*)))*)))*),)0(((-((((-.(4(,,))).,(())))))).)))))))-))-(It means that the fragment named @M00970 corresponds to
the DNA sequence
GTGCCAGCCGCCGCGGTAGTCCGACGTGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACCGAAGAACATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAAAAAACAAAAAAAAAAAAAGAAGCAAATGACGATTCAAGAAAGAAAAAAACACAGAATACTAACAATAAGTCATAAACATCATCAACATAAAAAAGGAAATACACTTACAACACATATCAATATCTAAAATAAATGATCAGCACACAACATGACGATTACCACACATGTGTACTACAAGTCAACTA
and this sequence has been sequenced with a quality
GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGFGGGGGGAFFGGFGGGGGGGGFGGGGGGGGGGGGGGFGGG+38+35*311*6,,31=******441+++0+0++0+*1*2++2++0*+*2*02*/***1*+++0+0++38++00++++++++++0+0+2++*+*+*+*+*****+0**+0**+***+)*.***1**//*)***)/)*)))*)))*),)0(((-((((-.(4(,,))).,(())))))).)))))))-))-(.
PHRED scores
THE FASTQ format encodes quality as ASCII values, which are mapped to quality scores in the PHRED format. The PHRED score is calculated from P, the probability of a base-calling error:
\(Q = -10\log{P}\)
These values are linked by the Phred Quality Score, Probability of incorrect base call, and Base call accuracy. We can summarise these:
PHRED Probability Accuracy
10 1 in 10 90%
20 1 in 100 99%
30 1 in 1000 99.9%
40 1 in 10,000 99.99%
50 1 in 100,000 99.999%
60 1 in 1,000,000 99.9999%FASTQE aids in the interpretation of these by mapping the Phred Quality Score and ASCII code to an Emoji:
PHRED ASCII Emoji
0 ! 🚫
1 ” ❌
2 # 👺
3 $ 💔
4 % 🙅
5 & 👾
6 ’ 👿
7 ( 💀
8 ) 👻
9 * 🙈
10 + 🙉
11 , 🙊
12 - 🐵
13 . 😿
14 / 😾
15 0 🙀
16 1 💣
17 2 🔥
18 3 😡
19 4 💩Much easier to understand! Here we show the emoji for Q scores below 20, which represents a 1 in 100 probability of a base error. Given there can be many thousands of reads and bases in even a small sequeuncing sample, the impact of these errors on downstream analysis and conclusions is significant. Steps we will see later in the lesson can be used to remove low quality reads.
Case Study
Let’s look at the output of FASTQE on our case study data:
which should give the following:
OUTPUT
$ fastqe /shared/fastqe/female_musk2.fastq.gz
Filename Statistic Quality
/shared/fastqe/female_musk2.fastq.gz mean 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😉 😉 😁 😁 😁 😁 😁 😉 😁 😁 😁 😁 😁 😁 😉 😁 😉 😁 😁 😉 😁 😁 😁
😁 😁 😛 😜 😉 😁 😉 😉 😁 😁 😁 😁 😁 😉 😁 😁 😁 😁 😁 😁 😁 😁 😉 😉 😉 😉 😉 😁
😁 😉 😁 😁 😉 😉 😉 😛 😋 😄 😄 😆 😄 😆 😆 😆 😆 😆 😘 😆 😆 😆 😆 😘 😘 😘 😘 😘
😘 😘 😘 😘 😘 😘 😘 😘 😘 😘 😘 😘 😃 😃 😘 😘 😘 😘 😘 😃 😃 😃 😃 😃 😃 😃 😘 😘
😃 😘 😃 😃 😘 😃 😃 😃 😃 😃 😃 😃 😃 😃 😃 😚 😃 😃 😃 😃 😃 😃 😃 😃 😃 😚 😃 😃
😃 😃 😃 😃 😃 😚 😚 😚 😚 😚 😃 😚 😚 😚 😚 😃 😚 😚 😃 😃 😚 😚 😚 😃 😚 😚 😚 😚
😚 😚 😚 😚 😚 😚 😚 😚 😚 😗 😚 😚 😚 😗 😗 😗 😗 😗 😚 😚 😗 😗 😗 😗 😗 😗 😙 😗
😙 😙 😙 😙 😗 😙 😏 😊 😙 😙 😙 😊 😊 😏 😊 😊 😊 😏 😏 😅 😏 😅 😏 😊 😊 😊 😅 😅
😅 😅 😏 😅 😏 😅 😊 😏 😅 😏 😏 😅 😅 😅 😅 😏 😅 😅 😏 😅 😅 😀 😀 😀 😅 😀 😅 😀
😅 😀 🚨 😡Challenge 5: What about the other sample?
Can you use fastqe on the other tissue sample? Which
sample has better overall quality? Why?
You should run fastqe with a different filename:
and the output will be:
OUTPUT
Filename Statistic Quality
/shared/fastqe/female_oral2.fastq.gz mean 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😁 😁
😁 😁 😉 😜 😛 😜 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😉 😁 😁 😉 😁 😁 😉 😉 😉 😉 😉
😉 😉 😉 😁 😉 😉 😉 😉 😛 😄 😘 😃 😃 😃 😚 😚 😚 😚 😚 😚 😗 😗 😚 😗 😗 😗 😗 😗
😙 😗 😗 😗 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙
😙 😙 😙 😙 😙 😙 😊 😙 😙 😙 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊
😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😏 😊 😊 😊 😏 😊 😏 😏 😏 😏 😊 😊 😊 😊 😊 😏 😏
😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😅 😅 😅 😅 😅 😅 😅
😅 😅 😅 😅 😅 😅 😀 😀 😀 😅 😀 😀 🚨 😀 😀 😀 😀 😀 😀 🚨 🚨 💩 🚨 🚨 😀 🚨 💩 💩
💩 🚨 🚨 🚨 🚨 💩 🚨 🚨 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩
💩 😡 💩 😾The mouth (oral) sample has inferirior quality, on average, towards the end of the reads.
To look at the quality of these files in more detail, we will next
use the emoji-less command line tool fastp.
Key Points
- The options available for
fastqe - The formula for Q scores
- FASTQE maps scores to emoji
- FASTQE can be used to quickly asses the quality of sequencing data
Content from Filtering with fastp
Last updated on 2024-09-08 | Edit this page
Filtering
The quality drop off observed could cause bias in downstream analyses with these potentially incorrectly called nucleotides. Sequences must be treated to reduce bias in downstream analysis. Trimming can help to increase the number of reads the aligner or assembler are able to succesfully use, reducing the number of reads that are unmapped or unassembled. In general, quality treatments include:
- Trimming/cutting/masking sequences:
- from low quality score regions
- beginning/end of sequence
- removing adapters
- Filtering of sequences
- with low mean quality score
- too short
- with too many ambiguous (N) bases
fastp is a command line tool that performs many of these
steps. Let’s run it with our oral2 sample, and direct output of the new,
filtered, data to a new file with -o opoption:
Challenge 6: What is filtered?
How many reads are in this FASTQ file before and after filtering?
After filtering, what is the % of reads with a Q score of 20 or higher?
429 reads are filtered, and 93.7328% of the remaining have a Q score of at leats 20.
OUTPUT
Read1 after filtering:
total reads: 383
total bases: 113368
Q20 bases: 106263(93.7328%)
Q30 bases: 98853(87.1966%)
Filtering result:
reads passed filter: 383
reads failed due to low quality: 428
reads failed due to too many N: 1
reads failed due to too short: 0
reads with adapter trimmed: 0
bases trimmed due to adapters: 0In addition to command line information, fastp produces
an interactive HTML report. We can use our browser platform to view
it:
Those reports help, but what about our old friend FASTQE? Recall the output on the original file:
OUTPUT
Filename Statistic Quality
/shared/fastqe/female_oral2.fastq.gz mean 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁
😁 😁 😁 😁 😁 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😁 😁
😁 😁 😉 😜 😛 😜 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😉 😁 😁 😉 😁 😁 😉 😉 😉 😉 😉
😉 😉 😉 😁 😉 😉 😉 😉 😛 😄 😘 😃 😃 😃 😚 😚 😚 😚 😚 😚 😗 😗 😚 😗 😗 😗 😗 😗
😙 😗 😗 😗 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙 😙
😙 😙 😙 😙 😙 😙 😊 😙 😙 😙 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊
😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😊 😏 😊 😊 😊 😏 😊 😏 😏 😏 😏 😊 😊 😊 😊 😊 😏 😏
😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😏 😅 😅 😅 😅 😅 😅 😅
😅 😅 😅 😅 😅 😅 😀 😀 😀 😅 😀 😀 🚨 😀 😀 😀 😀 😀 😀 🚨 🚨 💩 🚨 🚨 😀 🚨 💩 💩
💩 🚨 🚨 🚨 🚨 💩 🚨 🚨 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩 💩
💩 😡 💩 😾Lets now run FASTQE on our filtered data from the oral smaple:
We can see easily the effect of the filtering and trimming done by
fastp:
BASH
Filename Statistic Quality
female_oral_filtered.fastq.gz mean 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😁 😁 😁 😁
😁 😁 😉 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😁 😉
😉 😉 😉 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😉 😉 😁 😁 😉 😉 😉 😉 😁 😉 😁 😁
😁 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉
😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😜 😜 😜 😜 😉 😉 😜 😜 😜
😜 😜 😉 😜 😜 😉 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜
😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😛 😜 😜 😜 😜 😜 😜 😜 😜 😛 😛 😜 😜
😛 😛 😛 😛 😛 😛 😛 😛 😛 😛 😝 😛 😛 😛 😛 😛 😛 😝 😝 😝 😝 😝 ☺️ 😝 😝 😝 😝 😝
😝 ☺️ ☺️ 😋 😋 ☺️ ☺️ ☺️ 😋 😄 😋 😋 😋 😋 😄 😋 😄 😄 😘 😄 😄 😋 😋 😘 😘 😆 😄 😆 😆
😆 😆 😄 😆 😘 😘 😆 😘 😘 😘 😘 😆 😘 😘 😘 😘 😘 😘 😘 😘 😘 😃 😘 😘 😃 😃 😃 😡Things are looking better!
Conclusion
To sum up, you just looked at Illumina FASTQ data quality using Emoji output. You then filtered the low quality sequences in your FASTQ file and output before and after QC plots. Following filtering, you created a new Emoji output of your filter FASTQ file.
You did all of that on the command line, congrats!
Key Points
-
fastpcan be used to filter low quality reads from sequencing data - Filtering can improve the quality of data and downstream analysis
- Emoji can be useful!