Command line tools for quality control
Last updated on 2024-09-08 | Edit this page
Estimated time: 7 minutes
Overview
Questions
- How to perform quality control of NGS raw data?
Objectives
- Assess short reads FASTQ quality using
FASTQE - Compare reads before and after quality filtering with
fastp
Introduction
During sequencing, the nucleotide bases in a DNA or RNA sample (library) are determined by the sequencer. For each fragment in the library, a sequence is generated, also called a read, which is simply a succession of nucleotides.
Modern sequencing technologies can generate a massive number of sequence reads in a single experiment. However, no sequencing technology is perfect, and each instrument will generate different types and amount of errors, such as incorrect nucleotides being called. These wrongly called bases are due to the technical limitations of each sequencing platform.
Therefore, it is necessary to understand, identify and exclude error-types that may impact the interpretation of downstream analysis. Sequence quality control is therefore an essential first step in your analysis. Catching errors early saves time later on.
To take a look at sequence quality along all sequences, we can use FASTQE. It is an open-source tool that provides a simple and fun way to quality control raw sequence data and print them as emoji. You can use it to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis.
We will also use the tool fastp which can be used to
filter sequence data to remove low quality reads.
Inline instructor notes can help inform instructors of timing challenges associated with the lessons. They appear in the “Instructor View”
OUTPUT
fastqe 1.0Challenge 2: Can you do the same for
fastp? Hint: try -v as well as
--version
Callout
When using the command line code, lines that start with the # sign are typically text descriptors or instructions, not lines of code.
The # character is used to indicate comments. A line starting with # line doesn’t do anything, in this case it’s just telling you what the next line of code does which is navigate to your computer’s desktop - We will stick with this convention throughout the activity - Comments can occur anywhere in a line, anything after the # will be ignored
OUTPUT
$ fastp --version
fastp 0.20.1You should get the same result from using -v and
--version which are known as short and long options
respectivlely. Long options will typically (but not always) have a
corresonidng short option.
Command line structure
We have mentioned commands and options so far. The syntax is typically as follows:

OUTPUT
dev/ home/ proc/ shared/ tmp/This is the filesystem of our browser-based portal.
Case Study
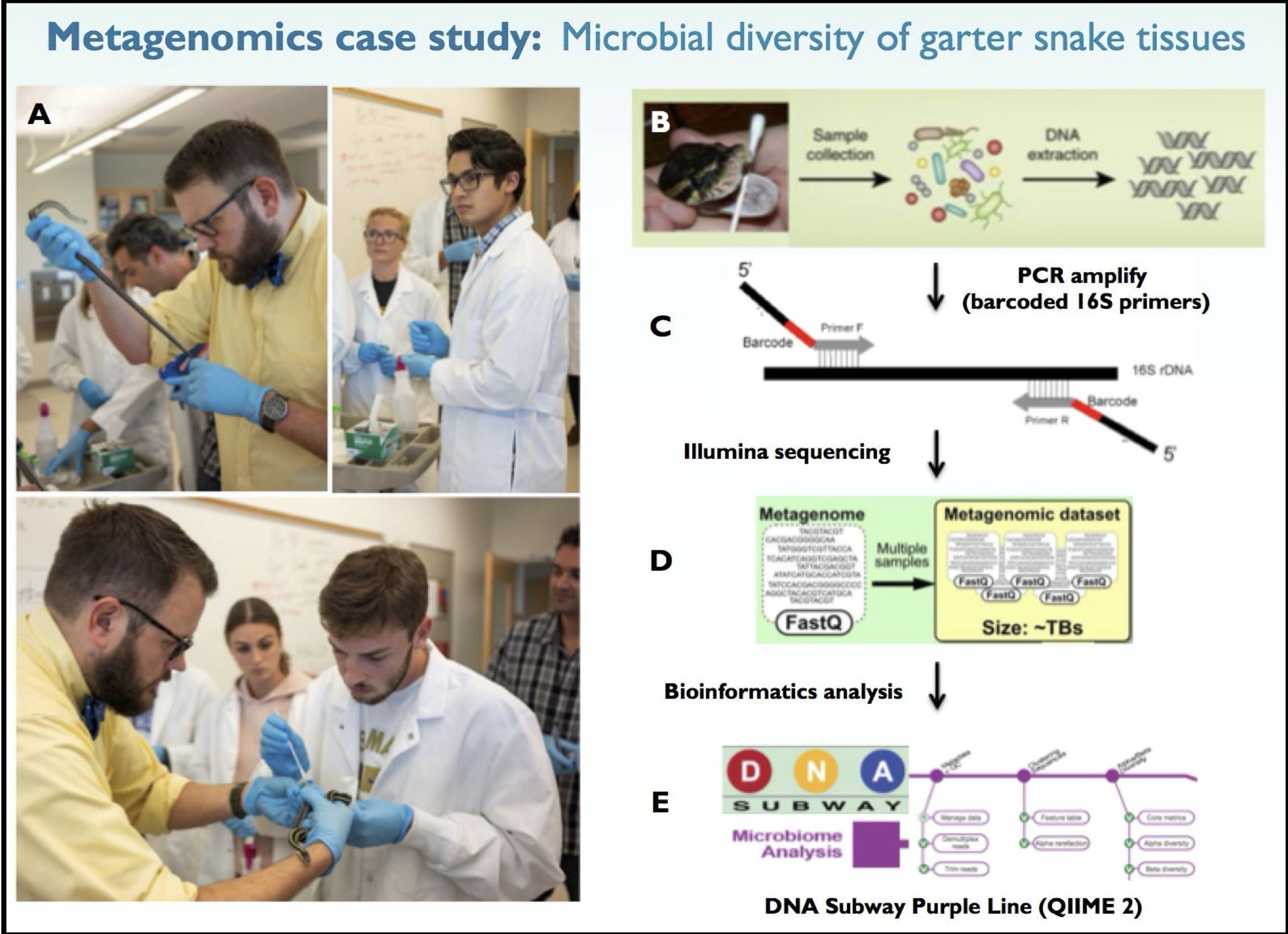
Here’s brief background on the in class metagenomics project that we’re collecting sequencing data for. Garter snakes excrete sexually dimorphic pheromones to attract a mate. The hypothesis of our experiment is that male and female garter snakes host unique microbial communities in their mouths, cloacae and musk glands that contribute to sexually dimorphic bioengineering of these pheromone molecules. Figure 1 provides an overview of our 16S metagenomics analysis pipeline. For this lesson though, all you need are the FASTQ files.
The data for this experiment is located in the
/shared/fastqe/ folder.
Challenge 4: What are the files available from this experiment?
Hint: Use the ls command we have already seen
Key Points
- Options can be long and short
- Structure of a shell command
- We have confirmed
fastpandfastqeare installed - The case study and location of the data